Quick Guide
Here you can find a short overview of all package modules as well as some main
applications with pyrexMD. Most functions are workflow-orientated so that you
can perform complex tasks (e.g. system setup, specific analyses, etc.) within a
few commands. This guide is kept very short because most commands have an
obvious ‘core’ functionality. All code snippets in this quick guide are taken
from the Jupyter notebooks within the example folder, which you can use to run
and test the package on your local machine.
Important
Most analysis functions for calculating useful quantities such as
RMSDs, Q values, contact distances, etc., can generate figures in the same
step if the keyword argument plot=True is passed. Please refer to the
API docs in order to get the most out of plot-generating functions, as they
usually allow many valid keyword arguments.
Module Overview
- pyrexMD.core
Contains functions enabling interactive analyses. Its main parts are the iPlayer and iPlot classes, which allow the use of a trajectory viewer or a dynamic linking of the trajectory viewer and any 2D graph.
- pyrexMD.gmx
Contains modified GromacsWrapper functions for streamlining the interaction with
GROMACSfor system setups etc.- pyrexMD.rex
Contains functions related to (contact-guided) Replica Exchange Molecular Dynamics, mainly for automating and speeding up the simulation setup.
- pyrexMD.topology
Contains functions for modifying universe topologies, e.g., align atoms/residues of two universes, get matching selection strings, include bias contacts.
- pyrexMD.analysis.analyze
Contains various functions for basic trajectory analyses, e.g., calculating RMSDs, distances, etc.
- pyrexMD.analysis.cluster
Contains functions for decoy clustering and post-REX clustering analyses.
- pyrexMD.analysis.contacts
Contains functions for native-contact and bias-contact analyses.
- pyrexMD.analysis.dihedrals
Contains functions for dihedral-angle analyses.
- pyrexMD.analysis.gdt
Contains functions for global distance test (GDT) analyses.
- pyrexMD.misc
Consists of pyrexMD.misc.classes, pyrexMD.misc.func, and pyrexMD.misc.plot. This sub-package is a collection of miscellaneous and frequently used functions and classes. These functions may contain modified versions of small existing functions to extend their default behavior in order to streamline
pyrexMD.
Application Overview
Setup of Normal MD Simulation
Using GROMACS in pyrexMD is very similar to the known command-line syntax.
Commands such as
gmx function -p parameter
simply become:
gmx.function(p=parameter)
Additionally to the expected GROMACS behavior, each gmx module function creates
by default a unique log file with a meaningful name which is stored in the
logs folder.
The code example below shows a complete setup of a normal MD simulation.
import pyrexMD.gmx as gmx
import pyrexMD.misc as misc
# create ref pdb:
pdb = "path/to/pdb"
ref = gmx.get_ref_structure(pdb, ff='amber99sb-ildn', water='tip3p', ignh=True)
# generate topology & box
gmx.pdb2gmx(f=ref, o="protein.gro", ff='amber99sb-ildn', water='tip3p', ignh=True)
gmx.editconf(f="protein.gro", o="box.gro", d=2.0, c=True, bt="cubic")
# copy mdp files (ions.mdp, min.mdp, nvt.mdp, npt.mdp, md.mdp) into working directory
misc.cp("path/to/mdp/files", ".")
# generate solvent & ions
gmx.solvate(cp="box.gro", o="solvent.gro")
gmx.grompp(f="ions.mdp", o="ions.tpr",c="solvent.gro")
gmx.genion(s="ions.tpr", o="ions.gro", neutral=True, input="SOL")
# minimize
gmx.grompp(f="min.mdp", o="min.tpr", c="ions.gro")
gmx.mdrun(deffnm="min")
# NVT equilibration
gmx.grompp(f="nvt.mdp", o="nvt.tpr", c="min.gro", r="min.gro")
gmx.mdrun(deffnm="nvt")
# NPT equilibration
gmx.grompp(f="npt.mdp", o="npt.tpr", c="nvt.gro", r="nvt.gro", t="nvt.cpt")
gmx.mdrun(deffnm="npt")
# MD run
gmx.grompp(f="md.mdp", o="traj.tpr", c="npt.gro", t="npt.cpt")
gmx.mdrun(deffn="traj")
Setup of Contact-Guided REX MD Simulation
The code example below shows a complete setup of a contact-guided REX MD simulation using different starting conformations (“decoys”) for each individual replica. It automates many system-specific and arduous tasks to eliminate possible application errors, such as mismatching system sizes across replicas, incorrect mapping of bias contacts, etc.
import pyrexMD.misc as misc
import pyrexMD.rex as rex
import pyrexMD.topology as top
decoy_dir = "path/to/decoy/directory"
# create rex_i directories and assign decoys
rex.assign_best_decoys(decoy_dir)
rex_dirs = rex.get_REX_DIRS()
# check for consistent topology
rex.check_REX_PDBS(decoy_dir)
# copy mdp files (ions.mdp, min.mdp, nvt.mdp, npt.mdp, rex.mdp) into working directory
misc.cp("path/to/mdp/files", ".")
# get parameters for fixed box size and solvent molecules
boxsize, maxsol = rex.WF_get_system_parameters(wdir="./rex_0_get_system_parameters/")
# create systems for each replica
rex.WF_REX_setup(rex_dirs=rex_dirs, boxsize=boxsize, maxsol=maxsol)
# minimize
rex.WF_REX_setup_energy_minimization(rex_dirs=rex_dirs, nsteps=10, verbose=False)
# add bias contacts (RES pairs defined in DCA_fin)
top.DCA_res2atom_mapping(ref_pdb=<ref_pdb>, DCA_fin=<file_path>, n_DCA=50, usecols=(0,1))
top.DCA_modify_topology(top_fin="topol.top", DCA_used_fin=<file_path> , k=10, save_as="topol_mod.top")
# prepare temperature distribution
rex.prep_REX_temps(T_0=280, n_REX=len(rex_dirs), k=0.006)
# create mdp and tpr files
rex.prep_REX_mdp(main_dir="./", n_REX=len(rex_dirs))
rex.prep_REX_tpr(main_dir="./", n_REX=len(rex_dirs))
# next: upload REX MD run files on HPC and execute production run
Interactive Plots
pyrexMD can generate interactive plots by linking a 2D graph to the trajectory
viewer of a specific universe. It allows to quickly inspect conformations at
specific values by interacting with the graph itself (e.g. via ctrl-click). In
this way, additional valuable information becomes accessible through the
trajectory viewer.
import MDAnalysis as mda
import pyrexMD.misc as misc
import pyrexMD.core as core
import pyrexMD.topology as top
import pyrexMD.analysis.analyze as ana
# set up universe
ref = mda.Universe(<pdb_file>)
mobile = mda.Universe(<tpr_file>, <xtc_file>)
# calculate RMSD
FRAMES, TIME, RMSD = ana.get_RMSD(mobile, ref=ref, sel1="protein", sel2="protein")
# create interactive plot
IP = core.iPlot(mobile, xdata=TIME, ydata=RMSD, ylabel=r"RMSD (A)")
IP()
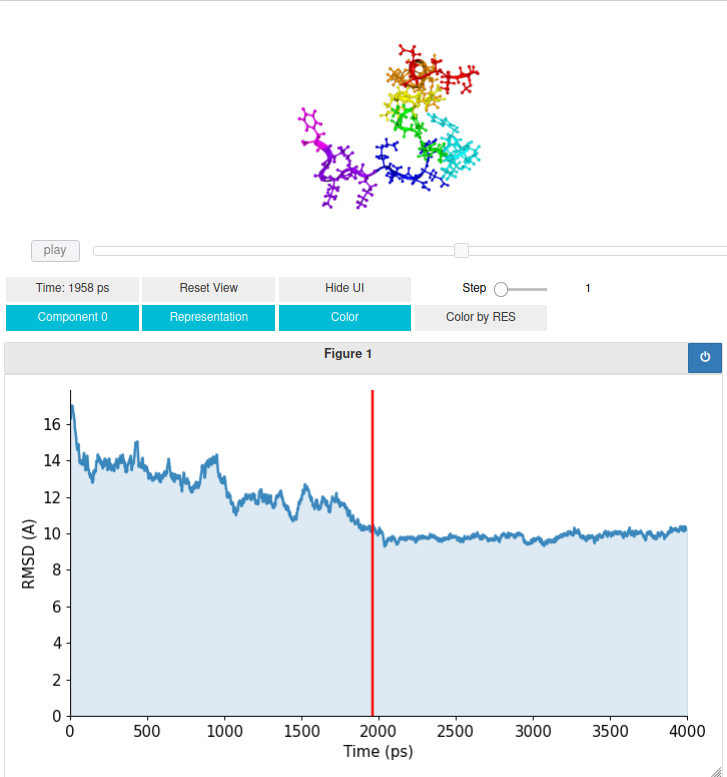
In this example, the interactive plot links the RMSD time evolution graph to the flexible trajectory.
Contact and Bias Analyses
REX is a very powerful and versatile sampling method. It improves sampling by
running many replicas in parallel over a wide temperature range and allows
switches of replicas between different temperatures while maintaining
thermodynamic ensembles. By integrating (theoretical, experimental, or mixed)
bias contacts via bias potentials, one can narrow down the search space and
guide the simulations towards specific conformations. This speeds up the process
and lowers the computational costs. pyrexMD covers many different forms of
contact and bias analyses.
import MDAnalysis as mda
import pyrexMD.misc as misc
import pyrexMD.topology as top
import pyrexMD.analysis.analyze as ana
import pyrexMD.analysis.contacts as con
# set up universes
folded = mda.Universe(<pdb_file_folded>)
unfolded = mda.Universe(<pdb_file_unfolded>)
mobile = mda.Universe(<tpr_file>, <xtc_file>)
top.norm_universe([folded, unfolded, mobile])
# check True Positive Rate (TPR) of predicted bias contacts
con.plot_DCA_TPR(folded, DCA_fin=<path_to_predicted_contacts>, n_DCA=80, d_cutoff=8.0)
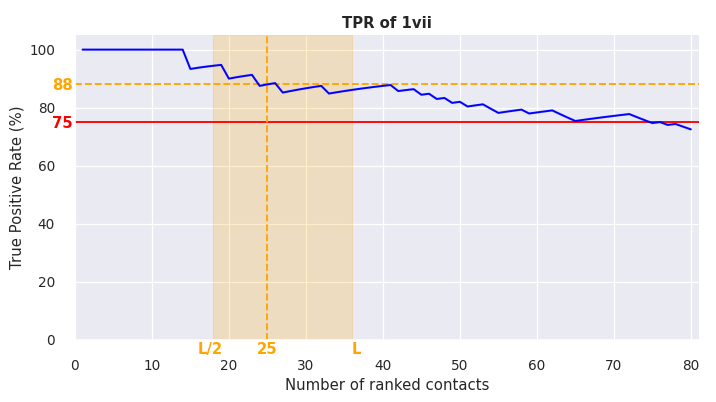
The figure shows:
blue line: TPR for number of ranked contacts
red line: 75% threshold (TPR of used contacts should be above approx. 75% for contact-guided REX MD, see https://doi.org/10.1371/journal.pone.0242072)
orange lines: suggested/guessed optimum number of contacts and the corresponding TPR
orange region: suggested region of interest between L/2 and L contacts (L = biomolecular sequence length)
# calculate QValue for realized bias contacts
bias_contacts = misc.read_file(<path_to_used_contacts>, usecols=(0,1))
FRAMES, QBIAS, CM = con.get_QBias(mobile, bc=bias_contacts)
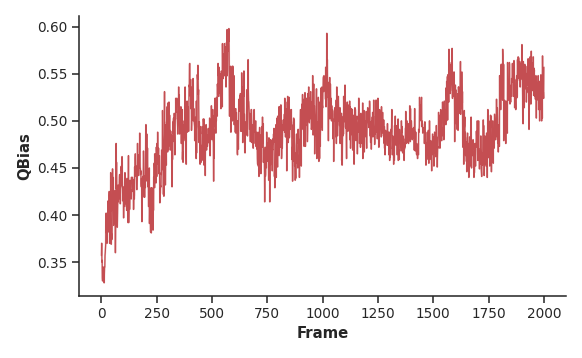
pyrexMD distinguishes mainly between two types of Q values, i.e., QNative
(fraction of native contacts) and QBias (fraction of realized bias contacts).
Both types can be used for structure analyses; however, when simulating unknown
target structures QNative becomes inaccessible due to the missing reference
structure.
# create log files with native contacts at different conformations
con.get_Native_Contacts(unfolded, sel="protein", save_as="unfolded_contacts.txt")
con.get_Native_Contacts(folded, sel="protein", save_as="folded_contacts.txt")
# check which contacts formed or broke up via ContactMap
fig, ax = con.plot_Contact_Map(folded, DCA_fin="unfolded_contacts.txt", sel="protein")
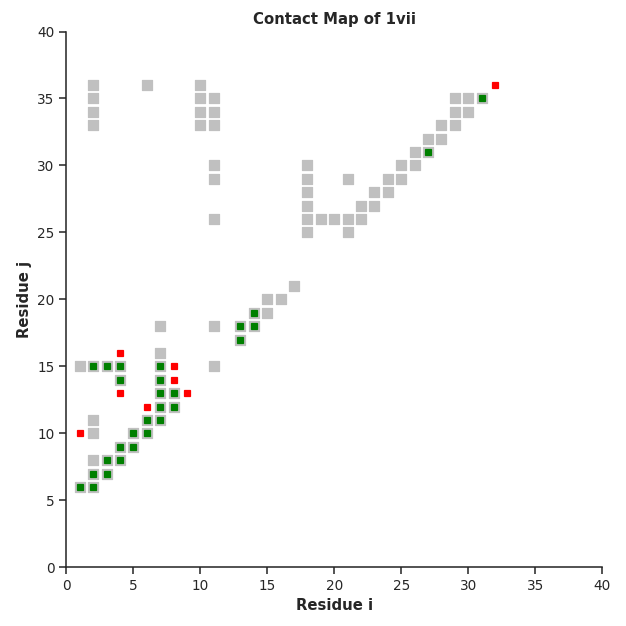
pyrexMD‘s Contact Maps show native contacts in grey and check whether bias
contacts are native (green) or non-native (red). This functionality can be used
to either compare and validate the used bias contacts or compare two structures
and show the newly formed and broken contacts in green and red, respectively.
# check native contact distances via ContactMap
NC, NC_dist, DM = con.get_NC_distances(folded, folded)
con.plot_Contact_Map_Distances(folded, NC=NC, NC_dist=NC_dist, sel="protein")
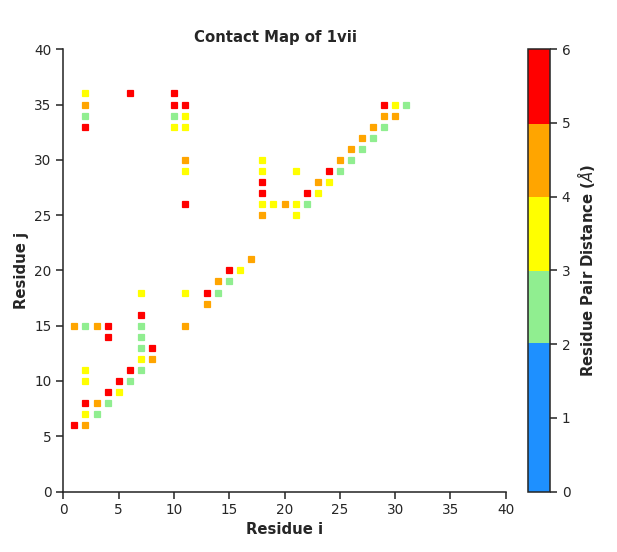
Additionally, it is possible to analyze native contact distances within a contact map plot. This can be used to deduce how strong/important the invidiual contacts are regards to structure stability.
GDT and LA Analyses
The so-called global distance test (GDT) is a method for structure evaluation similar to the root-mean-square deviation (RMSD). However, RMSD is a suboptimal measure of structural similarity as it strongly correlates with the largest displacement between mobile and target structure. If the mobile structure globally fits the target to a large extent and only one small segment is misaligned locally, the RMSD becomes disproportionately large. For the GDT, the mobile structure is first aligned to the target structure analogously to an RMSD analysis. To estimate how similar the two structures are, the displacement of each residual Cα atom is calculated and compared to various cutoffs. In a last step, percentages of residues with displacements below a considered threshold are used to calculate scores. The two most common scores are the total score (TS),
and the high-accuracy (HA) score,
where \(P_x\) denote the percentage of residues with displacements below a distance cutoff of x Å.
import MDAnalysis as mda
import pyrexMD.misc as misc
import pyrexMD.core as core
import pyrexMD.topology as top
import pyrexMD.analysis.analyze as ana
import pyrexMD.analysis.gdt as gdt
# set up universes
ref = mda.Universe("<pdb_file>")
mobile = mda.Universe("<tpr_file>", "<xtc_file>")
top.norm_and_align_universe(mobile, ref)
# perform GDT (Global Distance Test)
GDT = gdt.GDT(mobile, ref)
GDT_percent, GDT_resids, GDT_cutoff, RMSD, FRAME = GDT
# calculate GDT scores
GDT_TS = gdt.get_GDT_TS(GDT_percent)
GDT_HA = gdt.get_GDT_HA(GDT_percent)
# rank scores
SCORES = gdt.GDT_rank_scores(GDT_percent, ranking_order="GDT_TS", verbose=False)
GDT_TS_ranked, GDT_HA_ranked, GDT_ndx_ranked = SCORES
# generate plots
ana.PLOT(xdata=frames, ydata=GDT_TS, xlabel="Frame", ylabel="GDT TS")
ana.plot_hist(GDT_TS, n_bins=20, xlabel="GDT TS", ylabel="Counts")
# Local Accuracy plot
gdt.plot_LA(mobile, ref, GDT_TS_ranked, GDT_HA_ranked, GDT_ndx_ranked)
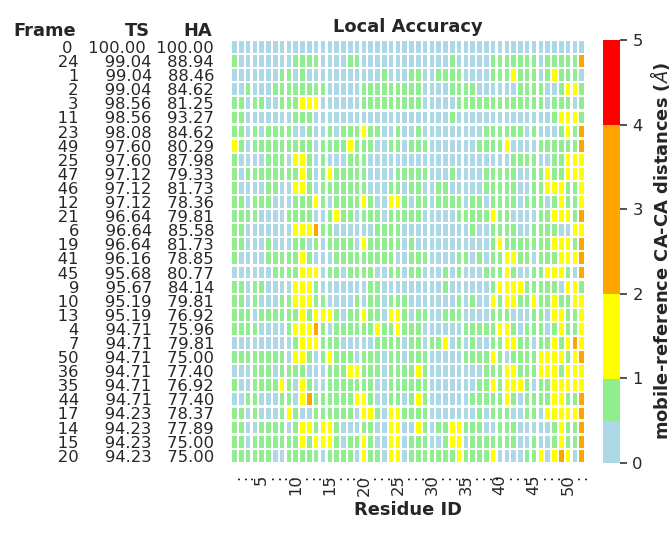
The local accuracy (LA) plot clearly shows how good each model part is refined compared to a reference structure. It is possible to show/hide each of the information columns (FRAME, TS and HA) individually.
Cluster Analyses
REX MD simulations generate large amounts of data. Depending on the project goal, filtering and clustering of structural ensembles will be necessary.
import pyrexMD.misc as misc
import pyrexMD.analysis.cluster as clu
# load data of pre-filtered frames
QDATA = misc.pickle_load("./data/QDATA.pickle")
RMSD = misc.pickle_load("./data/RMSD.pickle")
GDT_TS = misc.pickle_load("./data/GDT_TS.pickle")
score_file = "./data/energies.log"
ENERGY = misc.read_file(score_file, usecols=1, skiprows=1)
DM = clu.read_h5("./data/DM.h5")
# apply TSNE for dimension reduction
tsne = clu.apply_TSNE(DM, n_components=2, perplexity=50)
### apply KMeans on TSNE-transformed data (two variants with low and high cluster number)
# note: here we set the high number only to 20 because our sample is small with only 500 frames
cluster10 = clu.apply_KMEANS(tsne, n_clusters=10)
cluster20 = clu.apply_KMEANS(tsne, n_clusters=20)
### plot cluster data
# here: TSNE-transformed data with n_clusters = 20
# also: plot cluster centers with different colors
# - red dot: n10 centers
# - black dot: n20 centers
clu.plot_cluster_data(cluster20, tsne)
clu.plot_cluster_center(cluster10, marker="o", color="red", ms=20)
clu.plot_cluster_center(cluster20, marker="o", color="black")
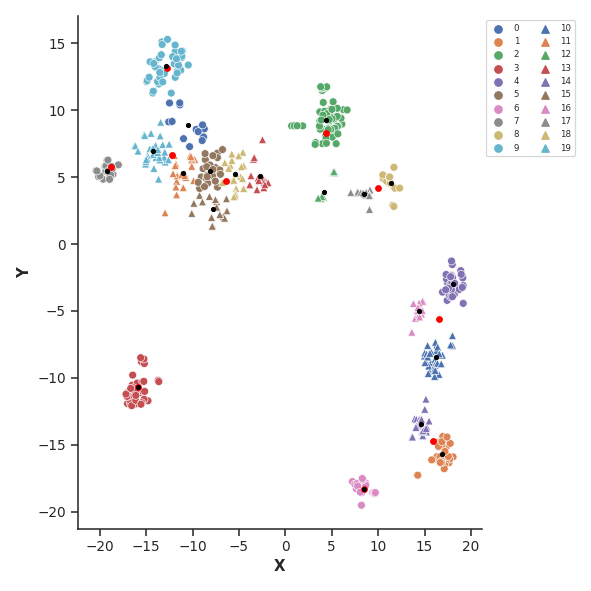
The example code above applies t-distributed stochastic neighbor embedding (TSNE) for dimension reduction of distance matrices (DM). Afterwards, a ‘fine’ and ‘coarse’ KMeans clustering is performed with 10 and 20 cluster centers, respectively.
It is possible to link both scores and structure accuracy to clusters, which can be used to select or compare the individual cluster ensembles, e.g. with
### map scores (energies) and accuracy (GDT, RMSD) to clusters
cluster10_scores = clu.map_cluster_scores(cluster_data=cluster10, score_data=score_file)
cluster10_accuracy = clu.map_cluster_accuracy(cluster_data=cluster10, GDT=GDT_TS, RMSD=RMSD)
cluster20_scores = clu.map_cluster_scores(cluster_data=cluster20, score_data=score_file)
cluster20_accuracy = clu.map_cluster_accuracy(cluster_data=cluster20, GDT=GDT_TS, RMSD=RMSD)
### print table with cluster scores stats
clu.WF_print_cluster_scores(cluster_data=cluster10, cluster_scores=cluster10_scores)
clu.WF_print_cluster_scores(cluster_data=cluster20, cluster_scores=cluster20_scores)
This prints a summary of the cluster scores (which can also be saved to a log
file if the save_as value is set).
cluster n10 scores (ranked by Emean)
ndx size compact | Emean Estd Emin Emax DELTA
6 77 6.695 |-230.652 6.975 -246.249 -211.738 -7.67
1 61 5.78 |-226.274 8.08 -241.86 -209.002 -3.292
8 43 3.098 |-225.174 7.679 -242.951 -206.42 -2.192
2 52 2.807 |-224.486 7.592 -240.913 -202.431 -1.504
7 41 9.439 |-223.741 17.481 -249.136 -190.634 -0.759
5 53 3.441 |-223.03 6.056 -237.002 -209.372 -0.048
9 25 2.172 |-220.319 7.431 -231.002 -203.796 2.663
0 80 9.121 |-216.962 7.09 -235.155 -200.969 6.02
3 25 0.798 |-214.371 6.688 -228.33 -201.657 8.611
4 43 1.91 |-194.022 2.585 -198.461 -190.412 28.96
-------------------------------------------------------------------
cluster n20 scores (ranked by Emean)
ndx size compact | Emean Estd Emin Emax DELTA
13 12 1.115 |-236.354 12.82 -249.136 -201.079 -13.372
12 22 2.137 |-231.452 5.878 -243.144 -219.911 -8.47
0 18 1.76 |-231.239 5.605 -246.249 -220.788 -8.257
8 14 0.497 |-230.679 11.003 -246.674 -208.399 -7.697
11 20 2.16 |-230.552 5.368 -242.131 -219.766 -7.57
15 31 1.638 |-229.789 8.104 -241.151 -211.738 -6.807
18 15 1.398 |-228.603 6.845 -241.86 -212.383 -5.621
5 43 3.098 |-225.174 7.679 -242.951 -206.42 -2.192
2 44 2.291 |-224.619 7.811 -240.913 -202.431 -1.637
19 8 0.384 |-223.752 6.2 -231.774 -211.164 -0.77
1 22 0.687 |-223.718 5.461 -236.263 -217.044 -0.736
17 15 0.448 |-222.686 6.776 -230.6 -205.661 0.296
14 31 1.334 |-222.541 6.4 -237.002 -209.372 0.441
10 36 2.35 |-222.276 8.934 -238.79 -203.796 0.706
4 21 0.526 |-221.485 7.025 -231.002 -207.53 1.497
9 33 1.409 |-217.024 6.648 -235.155 -204.817 5.958
7 25 0.798 |-214.371 6.688 -228.33 -201.657 8.611
6 32 0.946 |-214.215 5.974 -231.045 -200.969 8.767
16 15 0.894 |-207.389 12.28 -235.212 -190.634 15.593
3 43 1.91 |-194.022 2.585 -198.461 -190.412 28.96
Analogously, the cluster accuracy can be displayed with
### print table with cluster accuracy stats
clu.WF_print_cluster_accuracy(cluster_data=cluster10, cluster_accuracy=cluster10_accuracy)
clu.WF_print_cluster_accuracy(cluster_data=cluster20, cluster_accuracy=cluster20_accuracy)
and results in
cluster n10 accuracy (ranked by GDT mean)
| GDT GDT GDT GDT | RMSD RMSD RMSD RMSD
ndx size compact | mean std min max | mean std min max
2 52 2.807 | 77.296 1.815 73.81 80.953 | 2.555 0.154 2.182 3.076
6 77 6.695 | 77.003 2.451 63.69 82.44 | 2.804 0.096 2.62 3.154
1 61 5.78 | 75.943 2.325 71.728 82.142 | 2.85 0.096 2.567 3.03
8 43 3.098 | 74.821 2.017 70.538 79.763 | 2.895 0.096 2.696 3.19
7 41 9.439 | 73.374 14.68 41.37 94.94 | 2.873 1.192 0.996 6.501
9 25 2.172 | 68.941 2.312 65.177 74.407 | 3.091 0.104 2.796 3.221
0 80 9.121 | 64.695 3.943 55.057 74.703 | 3.444 0.238 2.719 3.896
5 53 3.441 | 63.235 1.766 58.927 66.668 | 3.498 0.132 3.079 3.721
3 25 0.798 | 60.012 2.289 56.248 63.69 | 3.804 0.087 3.684 4.043
4 43 1.91 | 55.621 2.013 51.785 60.715 | 4.312 0.17 3.794 4.798
---------------------------------------------------------------------------------
cluster n20 accuracy (ranked by GDT mean)
| GDT GDT GDT GDT | RMSD RMSD RMSD RMSD
ndx size compact | mean std min max | mean std min max
13 12 1.115 | 83.037 1.806 78.275 85.418 | 2.614 0.201 2.09 2.871
8 14 0.497 | 82.93 10.438 52.082 94.94 | 1.773 0.773 0.996 4.123
15 31 1.638 | 77.881 2.088 71.133 82.44 | 2.767 0.063 2.634 2.908
11 20 2.16 | 77.858 2.731 72.025 82.142 | 2.843 0.072 2.739 2.981
2 44 2.291 | 77.402 1.833 73.81 80.953 | 2.556 0.166 2.182 3.076
0 18 1.76 | 77.018 1.318 74.108 78.87 | 2.871 0.076 2.717 2.999
19 8 0.384 | 76.713 1.594 74.703 79.763 | 2.553 0.058 2.481 2.664
18 15 1.398 | 76.668 1.648 74.108 78.87 | 2.767 0.117 2.567 2.931
12 22 2.137 | 75.744 2.881 63.69 79.168 | 2.796 0.119 2.62 3.154
5 43 3.098 | 74.821 2.017 70.538 79.763 | 2.895 0.096 2.696 3.19
10 36 2.35 | 74.257 1.696 69.943 78.573 | 2.891 0.072 2.734 3.03
17 15 0.448 | 70.22 2.486 65.18 74.703 | 3.101 0.163 2.719 3.401
4 21 0.526 | 68.467 2.186 65.177 74.407 | 3.124 0.071 2.923 3.221
9 33 1.409 | 65.558 1.426 62.795 68.157 | 3.452 0.144 3.181 3.879
1 22 0.687 | 64.273 1.079 62.498 66.37 | 3.582 0.078 3.449 3.721
14 31 1.334 | 62.499 1.79 58.927 66.668 | 3.439 0.129 3.079 3.635
6 32 0.946 | 61.216 2.586 55.057 66.968 | 3.597 0.172 3.239 3.896
7 25 0.798 | 60.012 2.289 56.248 63.69 | 3.804 0.087 3.684 4.043
16 15 0.894 | 56.726 6.906 41.37 67.858 | 4.105 0.773 3.18 6.501
3 43 1.91 | 55.621 2.013 51.785 60.715 | 4.312 0.17 3.794 4.798